
Patologie dei surreni
Le patologie surrenaliche di interesse chirurgico si distinguono in malattie con un eccesso di produzione di ormoni e masse surrenaliche non funzionanti.
Iperaldosteronismo primitivo (Morbo di Conn)
L'iperaldosteronismo primitivo (chiamato anche morbo di Conn) è una patologia caratterizzata dall'eccessiva secrezione di aldosterone da parte di una o di entrambe le ghiandole surrenali. L'aggettivo "primitivo" indica che il problema è localizzato proprio a livello dei surreni; infatti, condizioni patologiche al di fuori dei surreni possono determinare un'eccessiva produzione di aldosterone, ma questa malattia è chiamata iperaldosteronismo secondario. Il morbo di Conn si riscontra preferibilmente in sogetti giovani adulti, con maggiore incidenza nel sesso femminile.
L'iperaldosteronismo primitivo può essere causato sia da patologie unilaterai (iperattività di una sola ghiandola surrenale) sia da patologie bilaterali (iperattività di entrambi i surreni). Le cause di iperaldosteronismo sono:
- adenoma unilaterale (75% dei pazienti);
- iperplasia uni o bilaterale (25% dei pazienti);
- carcinoma (<1% dei pazienti);
- iperaldosteronismo familiare di tipo 1 o di tipo 2 (<1% dei pazienti).
Il segno più frequente del morbo di Conn è l'ipertensione arteriosa resistente, che è difficile da controllare perchè non risponde alle normali terapie mediche e richiede una terapia con più farmaci antipertensivi. La ridotta concentrazione di potassio nel sangue (ipokalemia) e la bassa acidità del sangue (alcalosi metabolica) sono altri due segni comuni. A lungo termine l'iperaldosteronismo danneggia irreversibilmente le arterie e il cuore. I sintomi dell'iperaldosteronismo primitivo sono determinati dall'ipertensione e dall'ipokalemia e includono cefalea, vista offuscata, facile affaticabilità, crampi, debolezza muscolare e paralisi temporanea.
La diagnosi di morbo di Conn si avvale in prima istanza di esami bioumorali, che dimostrino elevati valori di aldosterone e ridotti valori di renina, i quali non variano a seconda della posizione, dell'assunzione di un carico di sodio o della somministrazione di captopril. Raggiunta la diagnosi, è necessario localizzare la sede della patologia e a tal fine si può ricorrere a TAC, RMN, scintigrafia con colesterolo marcato e AVS (cateterismo venoso selettivo delle vene surrenaliche).
La terapia medica prevede il controllo della pressione arteriosa con farmaci antipertensivi (spesso sono necessari più farmaci in combinazione) e la correzione della potassiemia con la somministrazione di potassio. La terapia chirurgica comporta una surrenectomia monolaterale, ovvero l'asportazione del surrene sede di patologia: l'intervento chirurgico permette una rapida normalizzazione della potassiemia, ma non sempre è in grado di garantire un'altrettanto efficace normalizzazione della pressione arteriosa, poichè nei pazienti con iperaldosteronismo primitivo di lunga durata il persistere per anni di elevati valori pressori causa un diffuso indurimento delle pareti delle arterie, inoltre, il morbo di Conn può non essere la sola causa dell'ipertensione arteriosa, ma può associarsi a un quadro di ipertensione essenziale (principalmente su base familiare). In ogni caso, in seguito all'intervento chirurgico, se il paziente non guarisce totalmente dall'ipertensione arteriosa, beneficia comunque di una riduzione dei farmaci antipertensivi e di una semplificazione dello schema terapeutico.
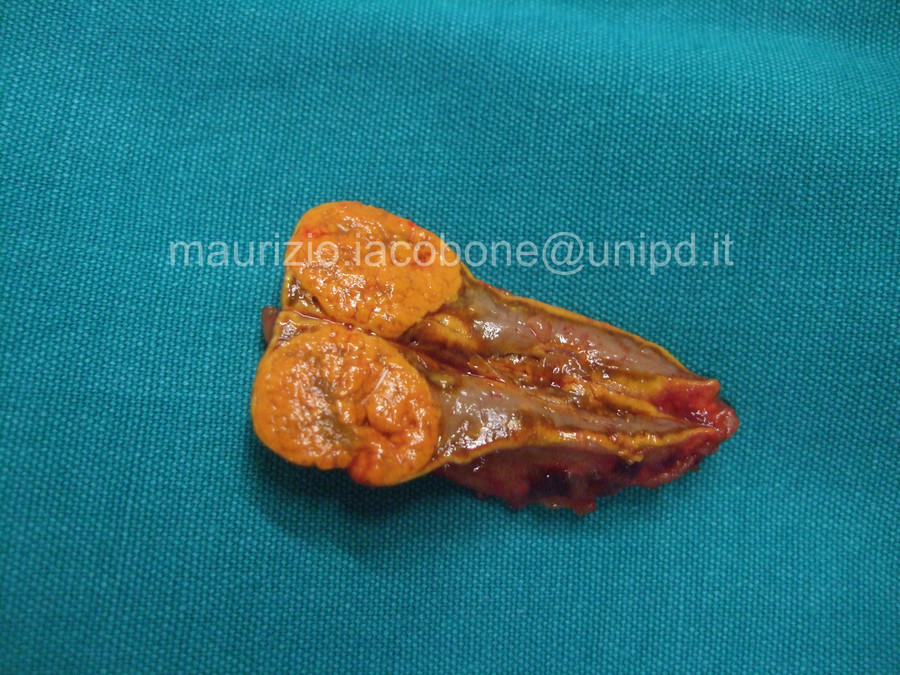 Adenoma surrenalico ipersecernente aldosterone.
Adenoma surrenalico ipersecernente aldosterone.
Ipercortisolismo (Sindrome di Cushing)
L'ipercortisolismo è una condizione patologica caratterizzata da un'iperproduzione di cortisolo. Si distingue:
- ipercortisolismo ACTH-indipendente = iperproduzione surrenalica di cortisolo, dovuta a una disfunzione primitiva del surrene (adenoma, iperplasia o carcinoma); rappresenta il 15-20% dei casi;
- ipercortisolismo ACTH-dipendente = iperproduzione surrenalica di cortisolo, dovuta a un'aumentata produzione di ACTH ipofisario (adenoma, microadenoma) o ectopico (sindrome paraneoplastica); rappresenta l'80-85% dei casi.
Segni e sintomi includono:
- aumento di peso, in particolare obesità a livello addominale;
- faccia "a luna piena", con forma arrotondata, talora con guance arrossate (pletora);
- gobba "di bufalo", alla base della nuca;
- strie viola (striae rubrae), alla base dell'addome;
- facilità alle escoriazioni e agli ematomi;
- cute sottile, "a carta velina" e difficoltà alla guarigione delle ferite;
- crescita anomala di peli in zone normalmente glabre, soprattutto nelle donne a livello del volto e dell'addome (ipertricosi);
- piedi e gambe gonfie (edema);
- ipertensione arteriosa;
- aumento della glicemia e diabete;
- debolezza muscolare e perdita del tono muscolare, con gambe e braccia sottili;
- alterazioni del ciclo mestruale (amenorrea) e diminuzione del desiderio sessuale;
- osteoporosi;
- aumentata suscettibilità allo sviluppo di trombosi (trombofilia);
- aumentata suscettibilità alle infezioni;
- cambiamenti dell'umore.
Alcuni di questi segni e sintomi non sono specifici dell'ipercortisolismo e possono presentarsi anche in altre condizioni patologiche. Pazienti che presentano un eccesso di cortisolo, ma non mostrano segni o sintomi di ipercortisolismo si definiscono essere affetti da ipercortisolismo subclinico.
La diagnosi si avvale di esami bioumorali specifici, quali il dosaggio dell'ACTH e del cortisolo nel sangue, il dosaggio del cortisolo nelle urine delle 24 ore, il dosaggio del cortisolo nella saliva e il dosaggio di ACTH e cortisolo nel sangue dopo somministrazione di desametasone (test di soppressione) e CRH (test di stimolo): questi esami permettono di stablire se l'ipercortisolismo è presente ed è ACTH-dipendente o indipendente. Una volta stabilita la diagnosi, è necessario localizzare la sede della patologia, attraverso TAC, RMN, scintigrafia con colesterolo marcato e sampling venoso selettivo.
La terapia medica della sindrome di Cushing prevede il trattamento dei segni e dei sintomi della malattia, per esempio la somministrazione di insulina per il diabete e farmaci antipertensivi per l'ipertensione arteriosa. Esistono pochi farmaci in grado di agire direttamente sull'iperproduzione di cortisolo: il mitotane distrugge le ghiandole surrenali e il ketoconazolo inibisce la produzione di cortisolo, ma sono entrambi gravati da importanti effetti collaterali. La terapia chirurgica comporta l'asportazione della ghiandola affetta (ipofisi o surrene) o della sede di produzione ectopica dell'ACTH. In caso di fallimento della terapia dell'ipercortisolismo ACTH-dipendente sulla sede di produzione dell'ACTH, può rendersi necessario asportare entrambi i surreni per controllare l'ipercortisolismo.
In generale, se la sindrome di Cushing non viene trattata adeguatamente, la patologia si aggrava progressivamente e i segni e i sintomi peggiorano: in particolare, il diabete e l'ipertensione possono causare seri problemi per la salute del paziente, fino all'ictus cerebrovascolare e all'infarto miocardico; anche la difficoltà alla guarigione delle ferite e la suscettibilità alle infezioni possono determinare gravi stati di sepsi.
 Faccia "a luna piena" con pletora.
Faccia "a luna piena" con pletora.
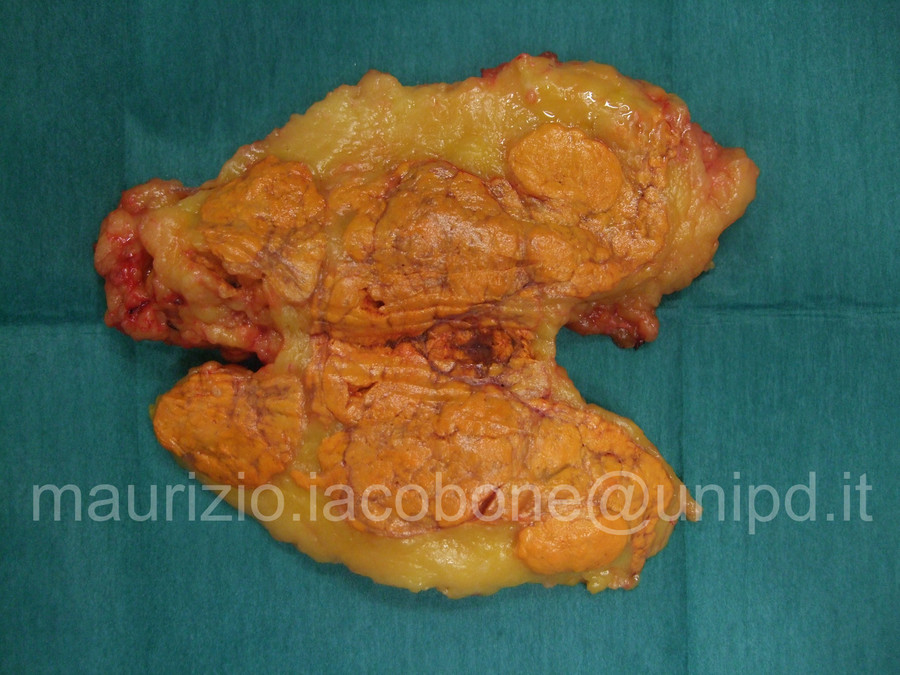 Surrene con iperplasia macronodulare ACTH-indipendente.
Surrene con iperplasia macronodulare ACTH-indipendente.
Feocromocitoma
Il feocromocitoma è una neoplasia che origina nella midollare del surrene e si caratterizza per un eccesso di produzione di catecolamine (adrenalina, noradrenalina e dopamina). Meno comunemente neoplasie simili possono svilupparsi anche al di fuori della ghiandola surrenale, a livello di strutture dette paragangli, e prendono il nome di PARAGANGLIOMI.
Il feocromocitoma può essere sporadico (senza alcuna familiarità evidente, 80-90% dei casi) oppure familiare (causato da una mutazione genetica ereditaria, 10-20% dei casi): le forme familiari più frequentemente delle forme sporadiche colpiscono entrambe le ghiandole surrenali e possono associarsi ad altre patologie endocrine e non.
Nella maggior parte dei casi, il feocromocitoma è benigno, ma in circa il 10% dei casi può essere maligno: è molto difficile distinguere un feocromocitoma benigno da uno maligno, poichè le sole caratteristiche che confermano la malignità sono la presenza di metastasi a distanza o di recidive locali di malattia.
Il sintomo principale è l'ipertensione arteriosa, che può presentarsi con pressione costantemente elevata o con picchi improvvisi di pressione alta (crisi ipertensive) o con una combinazione dei due. E' importante ricordare che non tutte le ipertensioni sono causate da feocromocitoma, ma meno dello 0,2% di ipertensioni all'anno sono riconducibili a un feocromocitoma. Altri sintomi includono: cefalea, vista offuscata, tachicardia, sudorazione, tremore, pallore, agitazione, iperglicemia.
La diagnosi si avvale del dosaggio delle catecolamine e dei loro metaboliti (le metanefrine) nelle urine delle 24 ore: tale analisi richiede di raccogliere le urine di un'intera giornata in un apposito contenitore, in cui è necessario versare un acido fornito al momento della consegna del contenitore stesso. Per identificare la sede del feocromocitoma si può ricorrere a TAC, RMN, scintigrafia con MIBG e DOPA-PET. In caso di sospetto di feocromocitoma è assolutamente controindicata la biopsia, la quale potrebbe scatenare una violenta crisi ipertensiva e causare gravi accidenti cardio e cerebrovascolari al paziente.
La terapia del feocromocitoma è in prima istanza chirurgica, attraverso l'asportazione della ghiandola surrenale patologica. La terapia medica è utile per preparare il paziente in vista dell'intervento chirurgico e prevede la somministrazione di farmaci che antagonizzano l'azione delle catecolamine (alfa-bloccanti eventualmente associati con beta-bloccanti). Non trattare adeguatamente un feocromocitoma è estremamente pericoloso e può condurre alla morte del paziente.
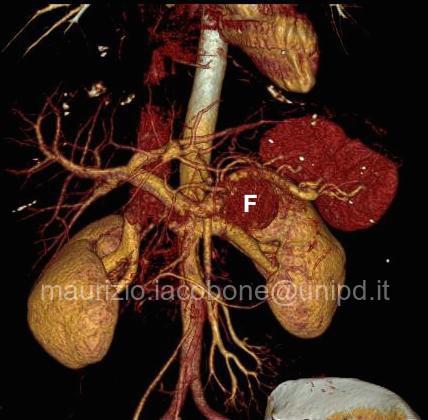 TAC con ricostruzione 3D di feocromocitoma sinistro (F).
TAC con ricostruzione 3D di feocromocitoma sinistro (F).
 Feocromocitoma sezionato.
Feocromocitoma sezionato.
Carcinoma corticosurrenalico
Il carcinoma corticosurrenalico è un tumore maligno raro, con circa 1-2 casi per milione di abitanti all’anno. Può presentarsi a tutte le età, ma colpisce soprattutto i bambini e gli adulti tra i 40 e i 50 anni. Il carcinoma corticosurrenalico è in genere sporadico (senza familiarità), ma esistono alcune rare forme familiari.
I carcinomi corticosurrenalici vengono spesso diagnosticati in fase avanzata, poiché causano segni e sintomi solo tardivamente, quando le dimensioni della massa sono tali da comprimere e dislocare le strutture circostanti. Possono essere presenti: affaticabilità e stanchezza (astenia), febbricola, perdita di peso, dolore addominale o lombare, gonfiore delle gambe (edema).
Nel 40% dei casi non si registra secrezione ormonale, mentre nel 60% dei casi il carcinoma è secernente (cioè è in grado di produrre ormoni) e spesso causa una sindrome da eccesso ormonale:
- ipercortisolismo (40% dei casi);
- virilizzazione (15-20% dei casi) = i soggetti di sesso femminile sviluppano peli in sedi tipicamente maschili (irsutismo), irregolarità mestruali, cambiamento del tono di voce, perdita del desiderio sessuale;
- femminilizzazione (5% dei casi) = i soggetti di sesso maschile sviluppano mammelle simili a quelle femminili (ginecomastia), perdita del desiderio sessuale;
- pubertà precoce = i bambini (sia maschi sia femmine) sviluppano segni di maturazione sessuale prima del normale.
La diagnosi deve essere sospettata in presenza di masse surrenaliche voluminose o a rapida crescita, ma spesso la diagnosi preoperatoria è difficile da confermare. Si può ricorrere a TAC, RMN e PET con 18FDG. La biopsia è assolutamente controindicata, perché può causare la rottura della capsula tumorale con possibile diffusione della malattia.
La terapia chirurgica con l’asportazione di tutta la massa in blocco con gli eventuali organi e strutture invase rappresenta la migliore possibilità di cura. Radio e chemioterapia sono scarsamente efficaci.
Il carcinoma corticosurrenalico è molto aggressivo: causa una diffusa invasione locale e metastasi a distanza (soprattutto al fegato e ai polmoni) e frequentemente recidiva dopo la chirurgia. La prognosi è infausta.
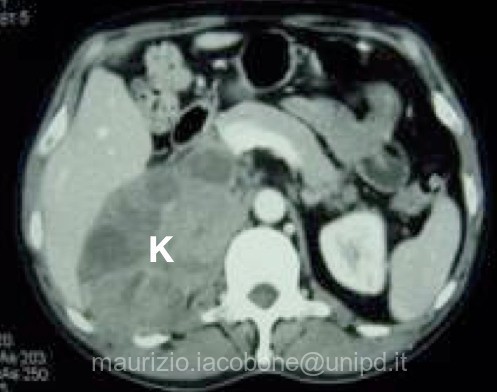 Carcinoma corticosurrenalico destro (K).
Carcinoma corticosurrenalico destro (K).
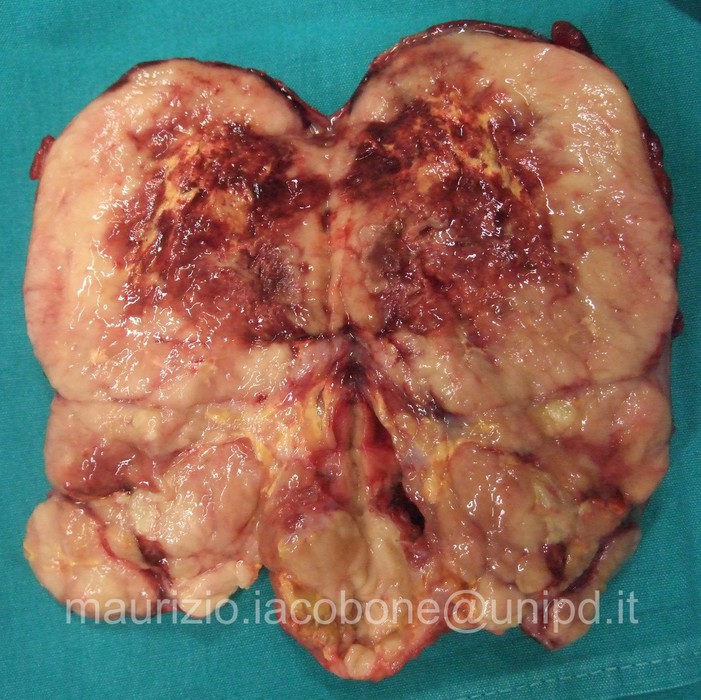 Carcinoma corticosurrenalico sezionato.
Carcinoma corticosurrenalico sezionato.
Metastasi surrenaliche
Le metastasi alle ghiandole surrenali sono frequenti e rappresentano la seconda lesione più comune dei surreni dopo gli adenomi benigni. Molteplici neoplasie possono localizzarsi secondariamente ai surreni: le più comuni sedi primitive sono polmone, mammella, colon, rene e melanoma.
Le metastasi alla ghiandola surrenale sono generalmente segno di una neoplasia ampiamente disseminata e spesso il coinvolgimento surrenalico è bilaterale. Le metastasi alla ghiandola surrenale sono in genere asintomatiche al momento della diagnosi. I sintomi più comunemente lamentati sono: senso di fastidio addominale, sensazione di tensione o peso al fianco e dolore alla schiena. Raramente causano insufficienza cortico-surrenalica.
La quasi totalità delle metastasi surrenaliche giunge all’osservazione del medico quale riscontro occasionale alle indagini radiologiche di follow-up oncologico. Talora l’esecuzione di un esame di imaging può essere suggerito da un rialzo inatteso dei marcatori tumorali.
I pazienti con lesioni metastatiche alla ghiandola surrenale richiedono una valutazione individualizzata da parte di un team multidisciplinare, al fine di stabilire l’iter terapeutico più appropriato per ciascun caso: in linea generale, pazienti con neoplasia largamente diffusa o in condizioni cliniche compromesse vengono indirizzati a chemioterapia o a cure palliative di supporto; viceversa, pazienti con malattia sensibile ad approcci più mirati e potenzialmente radicali sono candidati alla chirurgia o a nuove terapie radio-interventistiche.
 Metastasi surrenalica da carcinoma renale.
Metastasi surrenalica da carcinoma renale.
Incidentaloma surrenalico
Con il termine "incidentaloma" si definisce una massa surrenalica scoperta occasionalmente in corso di indagini svolte per altri motivi. Il riscontro di tali lesioni è divenuto più frequente in questi ultimi anni, perché sempre più pazienti si sottopongono a indagini radiologiche. E’ importante sottolineare che il termine "incidentaloma" non definisce né la benignità né la malignità di una massa surrenalica.
Una volta riscontrato un incidentaloma è necessario stabilire:
- se la massa produce un eccesso di ormoni = dosaggi ormonali plasmatici e urinari;
- se la massa è benigna o maligna = studio delle morfologia e dell’evoluzione nel tempo della massa (cresce o rimane stabile?), attraverso TAC, RMN, PET.
Oltre alle patologie già trattate in precedenza (morbo di Conn, sindrome di Cushing, feocromocitoma, carcinoma corticosurrenalico, metastasi surrenalica), altre masse surrenaliche possono essere adenomi non secernenti, mielolipomi (tumori benigni composti di tessuto adiposo e tessuto mieloide), cisti o altre neoplasie più rare.
Non tutti gli incidentalomi surrenalici richiedono un intervento chirurgico, ma la terapia chirurgica è indicata in caso di:
- segni morfologici di malignità (invasività locale);
- dimensioni superiori a 5 cm;
- rapida crescita;
- produzione di un eccesso di ormoni.
Qualora l’intervento chirurgico non sia indicato, si effettua un follow-up a 6-12 mesi.
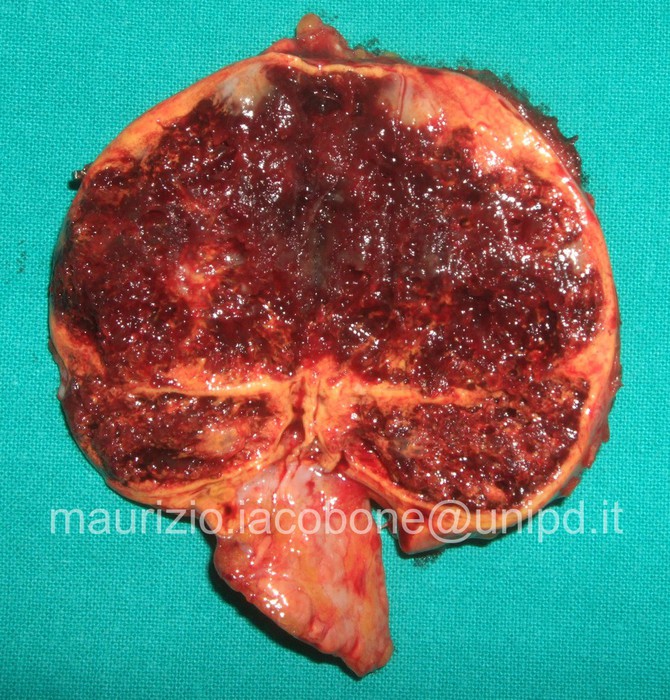 Emangioma surrenalico sezionato.
Emangioma surrenalico sezionato.