
Sindromi MEN (Multiple Endocrine Neoplasia)
Le sindromi MEN sono patologie endocrine molto rare, trasmesse come caratteri autosomici dominanti (è sufficiente che uno solo dei due geni sia mutato per causare la malattia), ma con espressività variabile (non tutti i soggetti con la mutazione hanno una sindrome identica). Sono caratterizzate dallo sviluppo di neoplasie in più ghiandole endocrine contemporaneamente o in tempi successivi: si presentano con associazioni costanti di patologie, legate a ben definite alterazioni genetiche.
La diagnosi di sindrome MEN è di estrema difficoltà e dovrebbe essere sospettata:
- in presenza di associazioni di almeno due componenti della sindrome;
- in presenza di una famiglia nella quale si siano manifestate in soggetti diversi singole componenti della sindrome;
- in caso di insorgenza in giovane età di una delle componenti della sindrome.
MEN1 (Sindrome di Wermer)
La sindrome MEN1 (Sindrome di Wermer) è costituita dall’associazione costante di:
- iperparatiroidismo primitivo;
- adenomi ipofisari;
- neoplasie del pancreas endocrino.
La sindrome MEN1 ha un’incidenza di 0,02/1000 e colpisce ugualmente maschi e femmine. In genere viene diagnosticata attorno a 40 anni di età. E’ causata dalla mutazione del gene MENIN, localizzato sul cromosoma 11.
L’IPT1 interessa oltre il 90% dei pazienti e rappresenta il più comune sintomo di esordio; in genere è dovuto a una patologia multighiandolare.
Gli adenomi ipofisari riguardano il 50-60% dei pazienti; si rendono manifesti con sintomi da compressione delle strutture vicine (soprattutto il chiasma ottico, con incapacità di percepire la metà esterna del campo visivo) e più raramente con iperproduzione ormonale (soprattutto prolattina e ormone della crescita).
Le neoplasie del pancreas endocrino colpiscono circa l’80% dei pazienti e presentano una notevole variabilità, potendo essere isolate o multiple, benigne o maligne; possono produrre un eccesso di ormoni, soprattutto gastrina e insulina.
La terapia chirurgica è rivolta verso le manifestazioni cliniche che, di volta in volta, si manifestano: l’IPT1 va trattato con paratiroidectomie totali (con autotrapianto) o subtotali, cui va sempre associata la timectomia; gli adenomi ipofisari sono meritevoli di terapia solo se sintomatici; le neoplasie del pancreas endocrino richiedono una particolare attenzione per la loro possibile malignità.
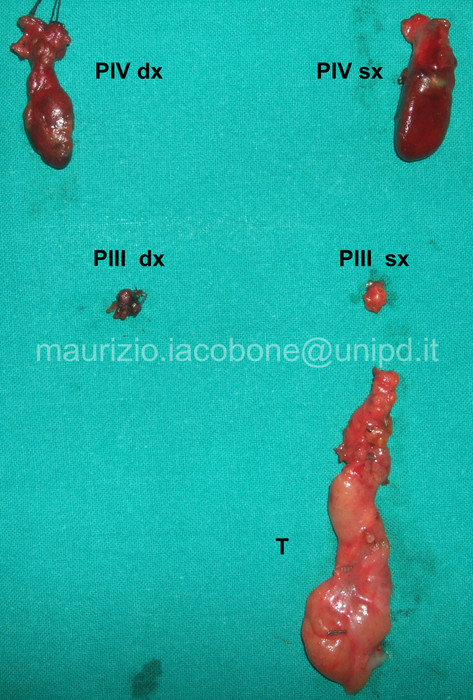 Paratiroidectomia subtotale (P) e timectomia (T).
Paratiroidectomia subtotale (P) e timectomia (T).
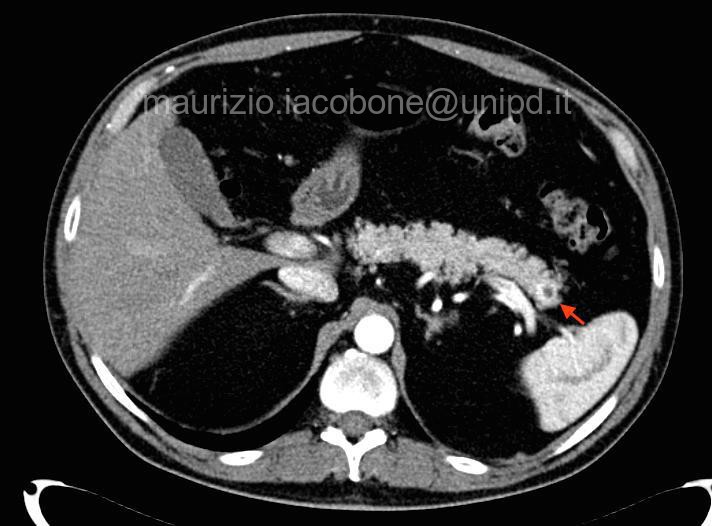 Neoplasia del pancreas endocrino (freccia rossa).
Neoplasia del pancreas endocrino (freccia rossa).
MEN2A (Sindrome di Sipple)
La sindrome MEN2A (Sindrome di Sipple) è costituita dall’associazione di:
- carcinoma midollare della tiroide;
- iperparatiroidismo primitivo;
- feocromocitoma.
E’ causata dalla mutazione del gene RET, localizzato sul cromosoma 10: in particolare la mutazione riguarda gli esoni 10 o 11 del gene RET.
Il carcinoma midollare della tiroide nella MEN2A costituisce il 10-15% di tutti i carcinomi midollari della tiroide; arriva a colpire il 100% dei portatori della mutazione e rappresenta la più comune patologia di esordio; l’età media di insorgenza è variabile, in genere entro i 20 anni di età; è quasi sempre multicentrico e associato a iperplasia delle cellule C; rappresenta la più frequente causa di morte per MEN2A (10% dei pazienti).
Il feocromocitoma colpisce circa il 50% dei pazienti; talvolta è bilaterale.
L’IPT1 interessa meno del 20% dei pazienti e in genere è dovuto a un adenoma singolo.
La terapia chirurgica, indipendentemente dalla patologia di esordio, non può prescindere dalla valutazione della presenza di feocromocitoma: in caso di presenza di feocromocitoma, infatti, la surrenectomia deve essere anteposta a qualsiasi altro intervento. La tiroidectomia totale è obbligatoria anche per i pazienti senza evidenza macroscopica di carcinoma midollare e deve essere condotta in via preventiva in età molto giovane (meno di 10 anni). L’IPT1 può essere trattato nella maggior parte dei casi con paratiroidectomia semplice.
MEN2B
La sindrome MEN2B è costituita dall’associazione di:
- carcinoma midollare della tiroide;
- feocromocitoma;
- habitus marfanoide;
- ganglioneuromatosi mucosa.
E’ causata dalla mutazione del gene RET, localizzato sul cromosoma 10: in particolare la mutazione riguarda l’esone 16 del gene RET.
Il carcinoma midollare della tiroide colpisce il 100% dei portatori della mutazione e rappresenta la più comune patologia di esordio; l’età media di insorgenza è precoce, in genere entro i 3 anni di età; è sempre multicentrico e associato a iperplasia delle cellule C.
Il feocromocitoma riguarda circa il 50% dei pazienti; talvolta è bilaterale.
L’habitus marfanoide interessa oltre l’80% dei pazienti e si caratterizza per alta statura, estremità (braccia e gambe) di lunghezza eccessiva rispetto al tronco, scarso sviluppo muscolare; possono associarsi altre deformità muscolo-scheletriche, quali pectus escavatum, cifosi, scoliosi, aumento della lassità dei legamenti, debolezza dei muscoli prossimali delle estremità.
La ganglioneuromatosi mucosa è descritta nella totalità dei pazienti ed è rappresentata dallo sviluppo di neurinomi mucosi multipli; tipica è la localizzazione alla mucosa della lingua, con il caratteristico aspetto della lingua "a lampone".
La terapia chirurgica, indipendentemente dalla patologia di esordio, non può prescindere dalla valutazione della presenza di feocromocitoma: in caso di presenza di feocromocitoma, infatti, la surrenectomia deve essere anteposta a qualsiasi altro intervento. La tiroidectomia totale è obbligatoria anche per i pazienti senza evidenza macroscopica di carcinoma midollare e deve essere condotta in via preventiva verso l’anno di vita.
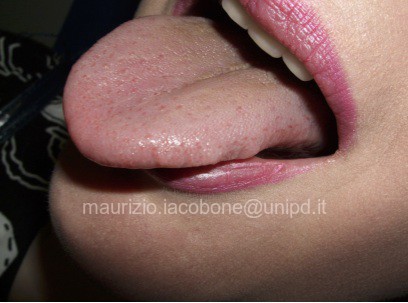 Ganglioneuromatosi mucosa: lingua "a lampone".
Ganglioneuromatosi mucosa: lingua "a lampone".
Carcinoma midollare tiroideo familiare
Anche se a rigore non è annoverabile tra le sindromi MEN, il carcinoma midollare tiroideo familiare (FMTC) condivide con le sindromi MEN2 il sito di mutazione, cioè il gene RET: in particolare, alla base dell'FMTC vi è la mutazione degli esoni 10, 11, 13 14 o 15 del gene RET.
Nei pazienti con FMTC il carcinoma midollare della tiroide è l'unica manifestazione clinica e non è associato ad altre neoplasie; l’età media di insorgenza è variabile, a seconda della mutazione; è quasi sempre multicentrico e associato a iperplasia delle cellule C.
La tiroidectomia totale è obbligatoria anche per i pazienti senza evidenza macroscopica di carcinoma midollare e deve essere condotta in via preventiva in età molto giovane.