
Hyperparathyroidism-Jaw Tumour Syndrome (HPT-JT)
L’iperparatiroidismo primitivo (IPT1) è una malattia endocrina comune, che si presenta nella maggior parte dei casi in forma sporadica; le forme ereditarie invece costituiscono meno del 20% dei casi e sono rappresentate soprattutto da sindromi MEN di tipo 1 e 2 e da Iperparatiroidismo Primitivo Familiare Isolato (in cui l’IPT1, pur avendo una distribuzione familiare, non è associato a altre manifestazioni fenotipiche: questa variante rappresenta un insieme eterogeneo di forme caratterizzate da mutazioni di CASR, ovvero da mutazioni non ancora identificate).
L’Hyperparathyroidism-Jaw Tumor Syndrome (HPT-JT) rappresenta un’ulteriore variante più rara, che si può manifestare con un IPT1 familiare non associato a MEN, con trasmissione autosomica dominante e penetranza incomplete (non tutti i soggetti con la mutazione sviluppano la malattia). E’ causata da mutazioni del gene CDC73 (o HRPT2), localizzato sul cromosoma 1 e codificante per la proteina parafibromina.
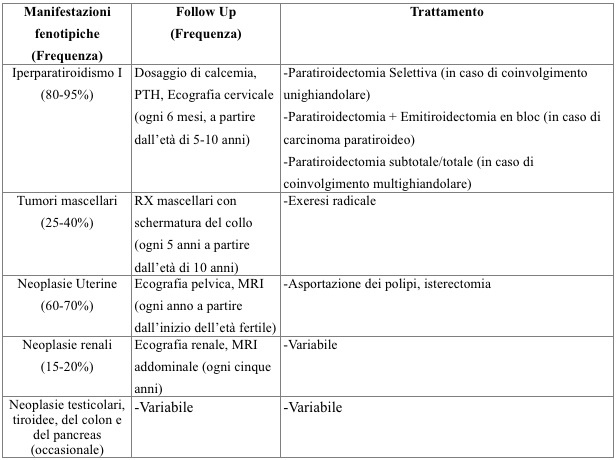
HPT-JT è caratterizzato dalla presenza di IPT1 (presente nell’80-95% dei casi), da neoplasie ossificanti dei mascellari (nel 25-40% dei casi), da coinvolgimento uterino (in circa il 60-70% delle donne portatrici della mutazione), da coinvolgimento renale (nel 15-20% dei casi). E’ stata inoltre segnalata la associazione con altre neoplasie: neoplasie germinali del testicolo (soprattutto seminomi), adenomi tiroidei a cellule di Hurthle, carcinomi tiroidei, adenocarcinoma del colon e lesioni pancreatiche.
Le indicazioni alla ricerca delle mutazioni di CDC73 sono:
- storia familiare suggestive per HPT-JT o Iperparatiroidismo familiare isolato (previa esclusione di mutazioni indicative di MEN1);
- IPT1 apparentemente sporadico ad insorgenza in età giovanile (< 35 anni);
- adenoma paratiroideo cistico;
- carcinoma paratiroideo;
- patologia paratiroidea multighiandolare;
- fibroma ossificante di mandibola o mascellare apparentemente sporadico in bambini.
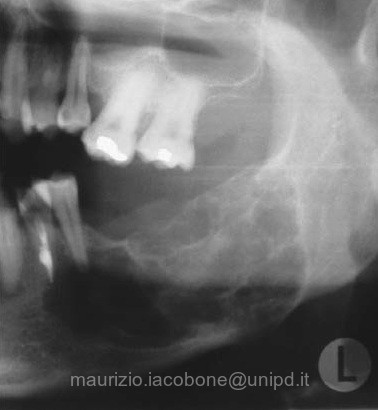 Fibroma ossificante del ramo sinistro della mandibola.
Fibroma ossificante del ramo sinistro della mandibola.
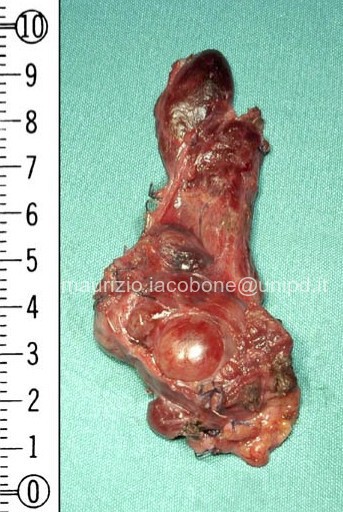 Paratiroidectomia e lobectomia tiroidea en bloc per carcinoma paratiroideo.
Paratiroidectomia e lobectomia tiroidea en bloc per carcinoma paratiroideo.