
Sindromi associate a feocromocitoma-paraganglioma
Con l’avanzamento delle conoscenze sulla genetica umana, si è scoperto che circa il 25-50% dei casi feocromocitoma sono dovuti a mutazioni genetiche. Le principali sindromi genetiche associate a feocromocitoma e paragangliomi sono quattro:
- SINDROME DI VON HIPPEL-LINDAU (VHL) = è causata da una mutazione del gene VHL, localizzato sul cromosoma 3: sono state descritte varie mutazioni del gene VHL. I pazienti con la sindrome di VHL possono sviluppare feocromocitomi (spesso bilaterali), paragangliomi, angiomi retinici (tumori degli occhi), emangioblastomi del sistema nervoso centrale (tumori dell’encefalo), carcinomi a cellule renali (tumori del rene), cisti pancreatiche e renali, tumori endocrini del pancreas e cistoadenomi dell’epididimo (tumori del testicolo). In alcune famiglie con sindrome di VHL il feocromocitoma o il paraganglioma è l’unico segno della malattia: complessivamente circa il 10-20% dei pazienti con sindrome di vHL sviluppa feocromocitoma o paraganglioma. Feocromocitomi bilaterali possono presentarsi in maniera sincrona (allo stesso momento da entrambi i lati) o separatamente un lato rispetto all’altro nel corso degli anni.
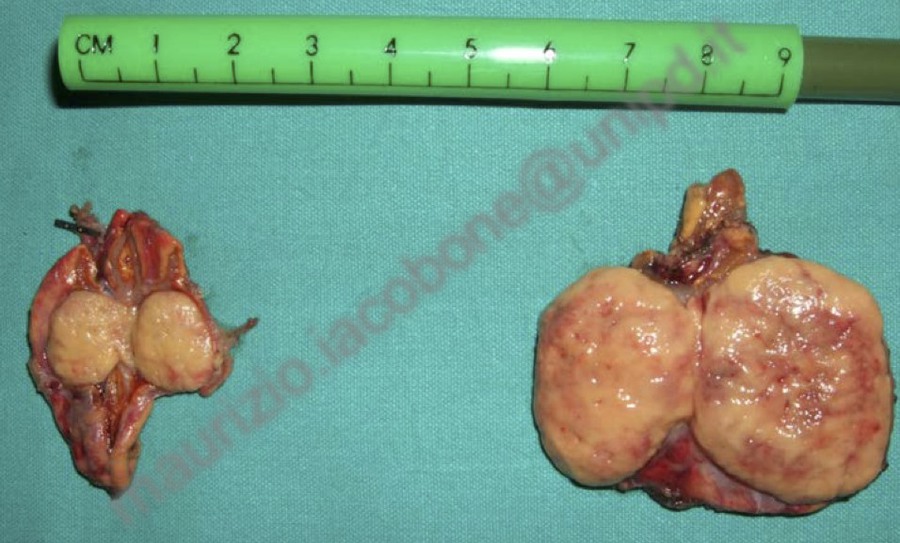 Feocromocitoma bilaterale in paziente di 9 anni con sindrome di vHL.
Feocromocitoma bilaterale in paziente di 9 anni con sindrome di vHL.
- NEUROFIBROMATOSI DI TIPO 1 (NF1) = è nota anche come sindrome di von Recklinghausen ed è causata dalla mutazione del gene NF1 localizzato sul cromosoma 17. I pazienti con NF1 presentano macchie color caffé-latte sulla cute e diffusi neurofibromi (tumori delle cellule nervose della pelle). Feocromocitomi o paragangliomi possono svilupparsi in oltre il 5% dei casi; raramente sono bilaterali.
- SINDROME DEL PARAGANGLIOMA FAMILIARE = è causata dalla mutazione della succinato-deidrogenasi (SDH): la mutazione della subunità B, C o D dell’enzima conduce a differenti forme della malattia.
Altre sindromi genetiche più rare associate a feocromocitoma e paraganglioma includono: la triade di Carney (tumori stromali gastrointestinali, condromi polmonari e paragangliomi) e le sindromi neurocutanee (atassia-telangectasia, sclerosi tuberosa, syndrome di Sturge-Weber). Anche mutazioni genetiche di TMEM-127 e MAX possono causare feocromocitoma.
La maggior parte di queste sindromi genetiche è autosomica dominante, cioè perchè si manifesti la malattia è sufficiente che sia mutata anche una sola delle due copie del gene: dunque i figli di un paziente con mutazione hanno il 50% di possibilità di sviluppare la stessa malattia del genitore.
Elementi di sospetto per un’origine genetica della patologia sono:
- diagnosi a un’età inferiore di 30 anni;
- feocromocitoma bilaterale;
- paraganglioma;
- storia familiare di feocromocitoma o paraganglioma;
- storia familiare delle sindromi descritte sopra;
- concomitanti segni e sintomi delle sindromi descritte sopra.